

 0
0 0
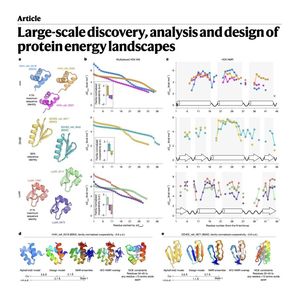
0This paper describes mHDX-MS, a new high-throughput experimental technique used to map the protein energy landscapes of thousands of small protein domains simultaneously. While traditional structural biology focuses on stable native states, this study identifies the rare conformational fluctuations and higher-energy states that dictate how proteins function and aggregate. By analyzing over 5,000 domains, the researchers discovered significant variations in opening cooperativity, proving that proteins with identical folds can exhibit vastly different local stabilities. The authors utilized Bayesian inference and machine learning to process the massive dataset, successfully identifying structural features that influence these movements. Ultimately, the study demonstrates that these data-driven insights allow for the precise design of mutations to stabilize specific structural segments. This multiplexed approach provides a transformative resource for improving artificial intelligence models used in protein engineering and biophysics.
References:
Ferrari Á J R, Dixit S M, Thibeault J, et al. Large-scale discovery, analysis and design of protein energy landscapes[J]. Nature, 2026: 1-11.
